Przypadek
1. Preparat Nr 3633
|
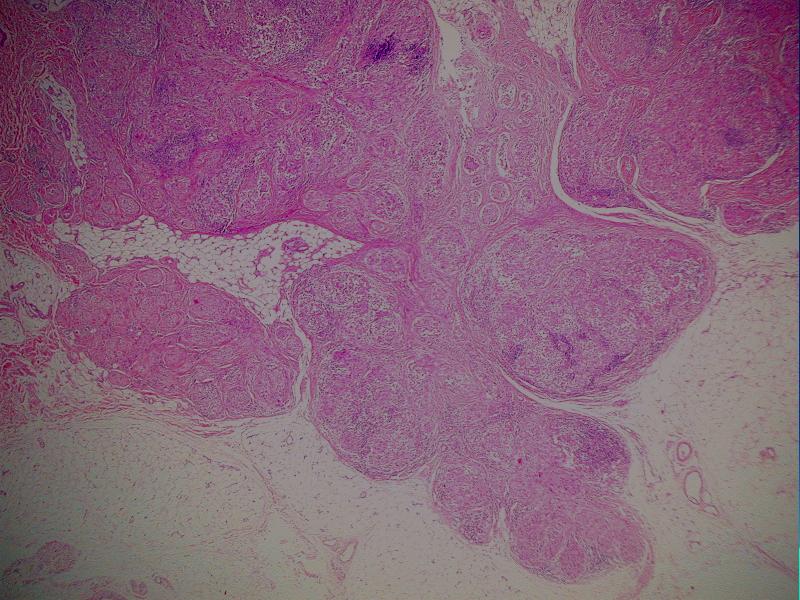
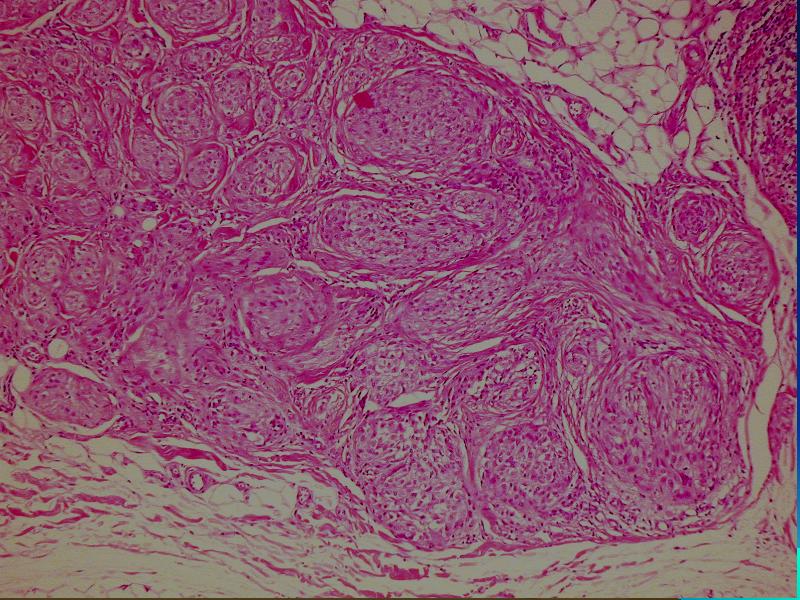
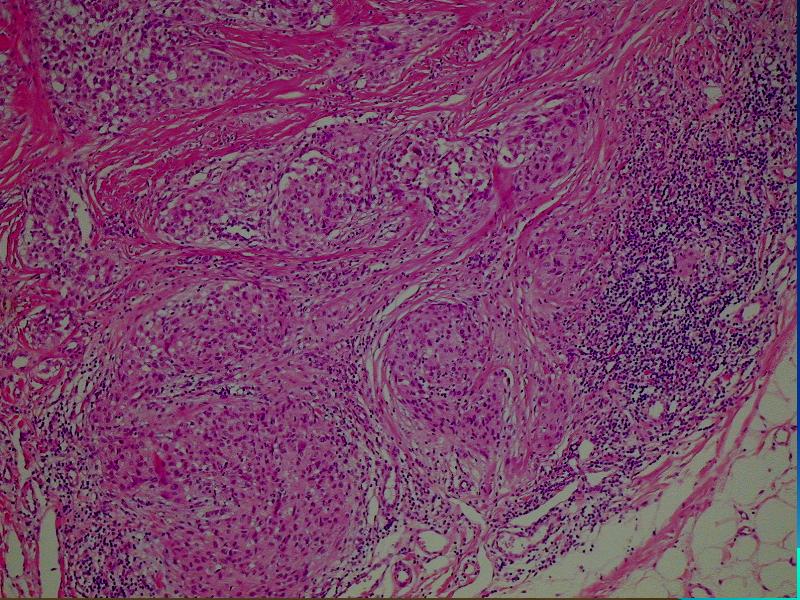
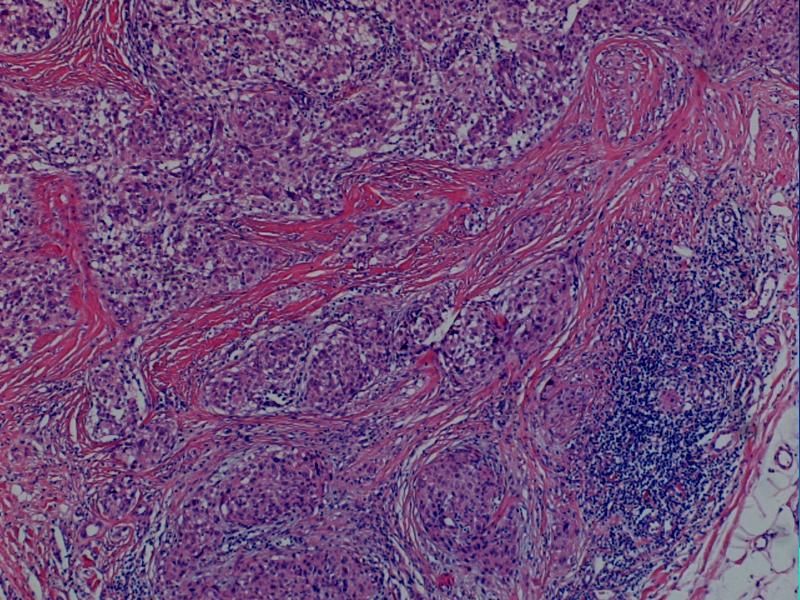
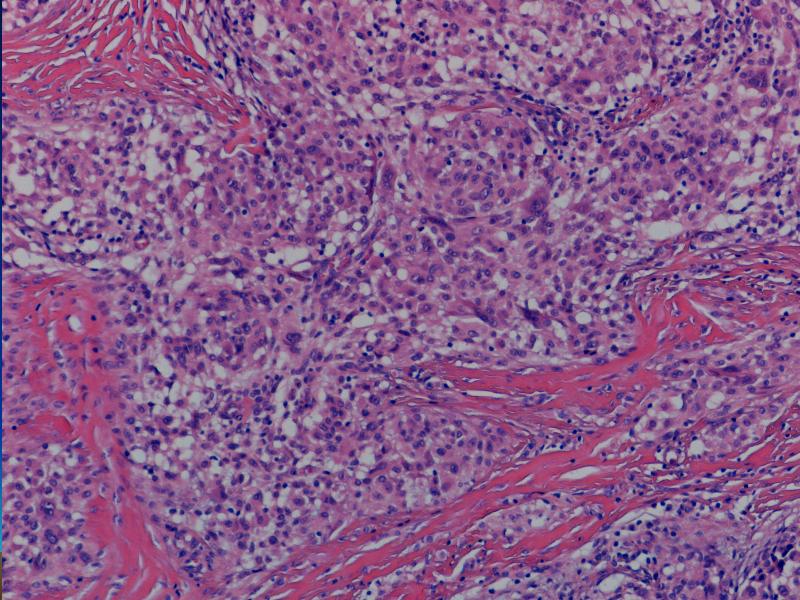
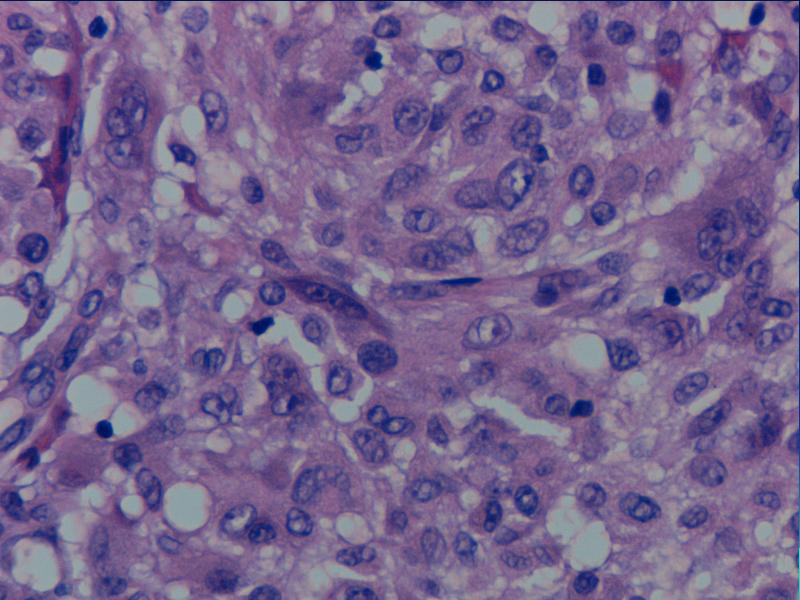
i Danuta Bręborowicz Ryhov Sjukhuset, Jonkoping, Szwecja. Dziewczynka 6 lat. W skórze zmiana tuż poniżej prawej pachwiny określona przez klinicystę jako ciało obce prawej pachwiny. Makroskopowo: Wycinek skóry z tkanką podskórną (16 x 7 x 8 mm). Opis pierwotny: w skórze właściwej /środkowa i dolna część/ obecny ziarniniak zbudowany ze zrazików zawierających komórki z obfitą cytoplazmą z pęcherzykowatymi jądrami z wyraźną błoną jądrową. Pojedyncze wielojądrowe komórki. Wokół zrazików i wewnątrz nich obecne limfocyty. Zmiana obecna również w tkance podskórnej. Usunięcie nieradykalne. Badanie immunohistochemiczne: vim +, część komórek CD68 +, S-100 -, CD31 -, keratin -. Nie stwierdzono ciał obcych podwójnie załamujących światło. Pierwotna diagnoza (E.N.) Podejrzenie sarkoidozy. Po trzech latach obserwacji i badań w kierunku sarkoidozy (wielokrotne Rtg płuc, badanie oczu) nie stwierdzono żadnych objawów sarkoidozy. Pediatra opiekujący się dzieckiem zażądał powtórnej oceny wykonaną przez FP. WYNIK: plexiform fibrohistiocytic tumor. W związku ze zmianą rozpoznania, klinicyści poprosili o konsultację ekspertów w zakresie guzów tkanek miękkich – Jeanne i Larsa-Gunnara Kndbloma. Oboje potwierdzili rozpoznanie FP. Wskazując na charakterystyczny sposób wzrostu jak i skład komórkowy. Jedynie obecność limfocytów w części peryferyjnej zrazików odbiega od klasycznego obrazu i mogą być powodem pomyłki diagnostycznej (sarkoidoza). Guz daje nawroty, jeśli nie jest radykalnie usunięty. Pediatra opiekujący się dzieckiem zażądał powtórnej oceny wykonaną przez FP. PLEXIFORM FIBROHISTIOCYTIC TUMOR – Guz występujący niemal wyłącznie u dzieci i młodych dorosłych, częściej u dziewczynek. Po raz pierwszy opisany przez Enzingera i Zhanga w 1988 r. Zlokalizowany w skórze w głębokich warstwach i w tkance podskórnej górnej kończyny (ponad 60% przypadków), kończyny dolnej (14%) i rzadziej w obrębie tułowia, głowy i karku. Wielkość 1-3cm, wyjątkowo>5cm. Słabo odgraniczony od otoczenia, spoisty, szarobiaławy. W badaniu mikroskopowym typowych przypadków (40%) stwierdza się dwa komponenty: fibroblastyczny i histiocytarny z obecnością komórek olbrzymich wielojądrowych. Komórki histiocytarne tworzą drobne guzki otoczone wiązkami komórek fibroblastycznych. Komórki wielojądrowe rozrzucone są nieregularnie wśród wyżej wspomnianych struktur. Mitozy nieliczne. Niekiedy ogniskowa atopia. Miejscami wylewy krwawe i złogi hemosyderyny i skupiska limfocytów. Fibroblasty tworzą krótkie wiązki oplatając guzki, tworząc splotowaty układ, który może upodabniać zmianę do fibromatozy. RÓŻNICOWANIE: Inflammatio granulomatosa np.: sarkoidoza. Dermatofibroma. Tumor gigantocellularis. Fibromatosis. Plexiform fibrohistiocytic tumor jest zaliczany do nowotworów o niskiej złośliwości, ze zdolnością do dawania nawrotów i wyjątkowo rzadko przerzutów (węzły chłonne, płuca). LECZENIE: Wycięcie w granicach tkanek zdrowych. Nie zaleca się adjuwantowej terapii z uwagi na małe ryzyko przerzutów i miejscowej wznowy. PIŚMIENNICTWO: Enzinger F.M. Zhang R: Plexiform fibrohistiocytic tumor presenting in children and young adults. An analysis of 65 cases. Am.J.Surg.Pathol. 1988 12:818-826 Coffin CM. Dehner LP, Meis-Kindblom JM: Inflammatory myofibroblastic tumor, inflammatory fibrosarcoma and reatet lesion: An historical review with differential diagnostic consideration. Semin Diagn. Pathol. 1998; 15:102-110 Miettinen M: Diagnostic Soft Tissue Pathology Chapter 6, p.181, 2003 Churchill Livingstone S.W.Weiss, J.R.Goldblum: Entzinger and Weiss`s Soft Tissue Tumors Chapter14, pp 522-527, 2001 Mosby. |